Fitness prediction across scales
Can a phylogenetic model predict the fitness effects of mutations in extant mammals?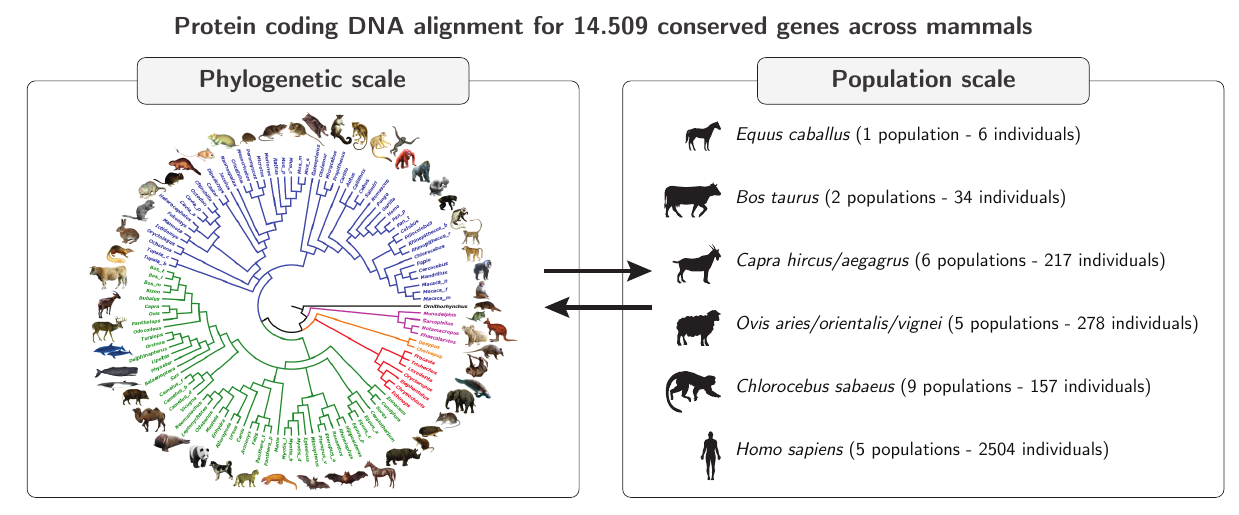
In this work based on genome-wide studies across mammalian species and populations, we estimated the proportion of beneficial mutations in protein coding sequences that are restoring pre-existing functions.
Our study is based on the premise that slightly deleterious mutations scattered across the genome are reaching fixation due to genetic drift.
These mutations are then subsequently reverted by beneficial back-mutations, generating a balance at which genomes are constantly both “damaged” and “repaired” simultaneously at different loci.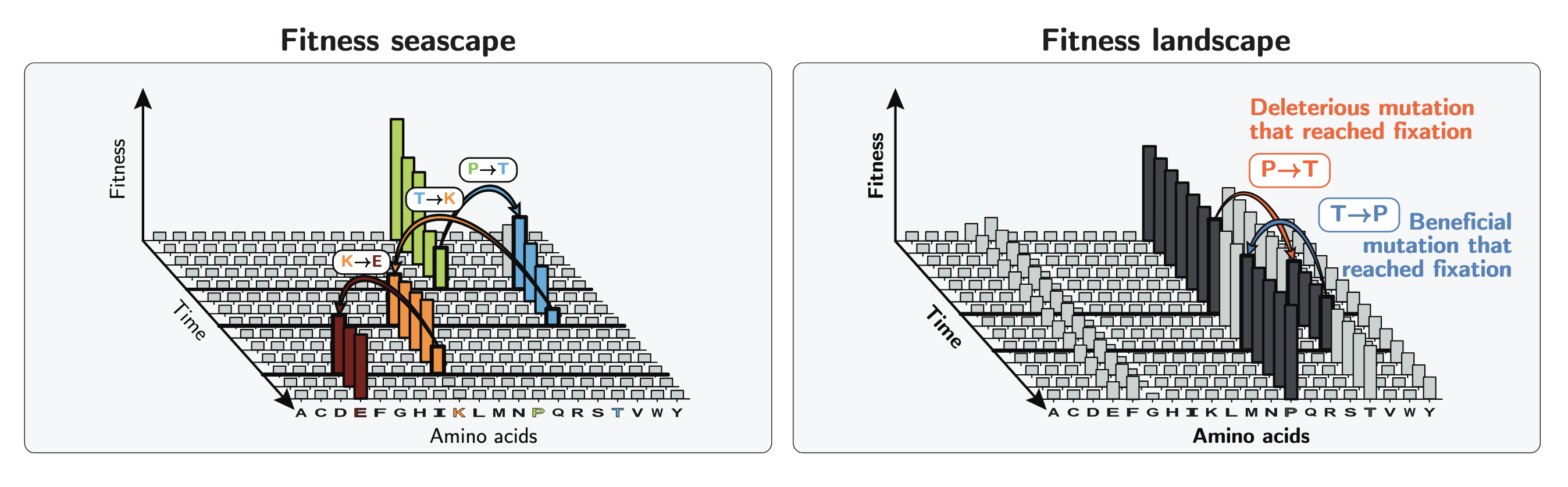
Even though the existence of these back-mutations is predicted by the nearly neutral theory, they have been largely overlooked, and positive selection has often been interpreted as adaptation to changing environments. Despite leaving similar signatures when looking only at populations, adaptive mutations promote phenotypic diversification while beneficial back-mutations reduce diversity between species and stabilize existing systems.
We first estimated selective effects of mutations inside mammalian protein coding sequences, under a model assuming no adaptation at the phylogenetic scale (see notes on Mutation-Selection models).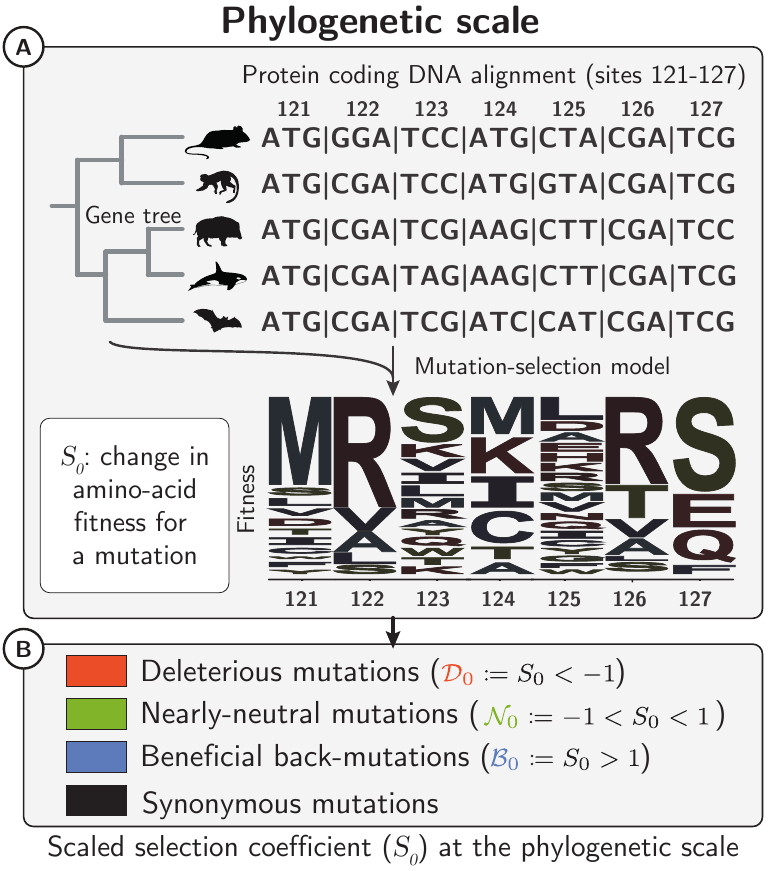
From these estimates, we then tested whether the fitness effects of mutations estimated at the phylogenetic scale could predict the fitness effects of mutations in extant populations.
We divided any mutations into three categories based on their predicted fitness effects: deleterious, nearly-neutral, and beneficial.
For each category, the fitness effects of non-synonymous mutations in populations were estimated from the site frequency spectrum of non-synonymous and synonymous polymorphisms.
The predicted fitness effects of mutations at the phylogenetic scale can then be compared to the fitness effects of mutations in populations.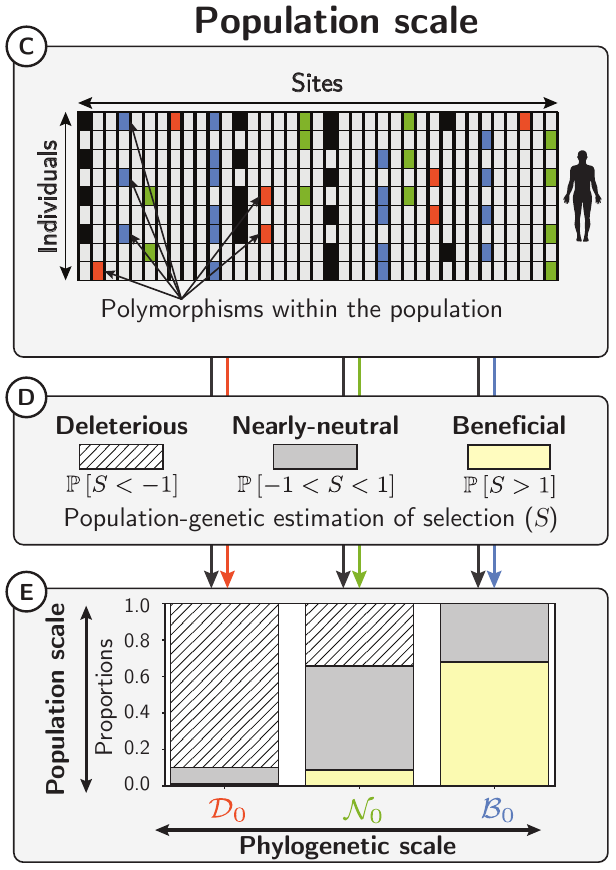
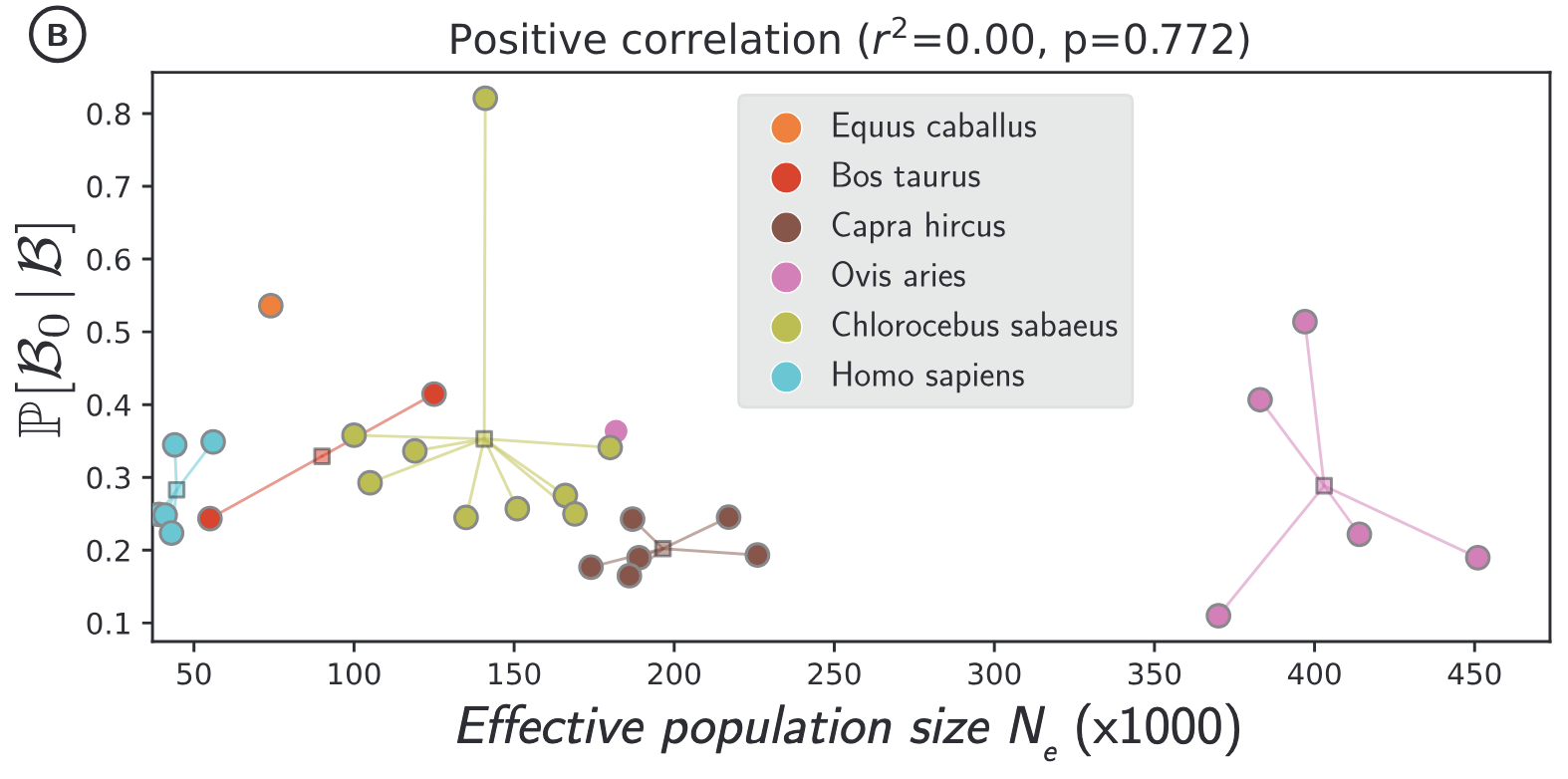
Since estimates at the phylogenetic scale are based on the assumption that no adaptation has occurred, using Bayes formula, we then estimated the proportion of beneficial mutations that are not adaptive innovations among all beneficial mutations at the population scale (P[B₀|B]).
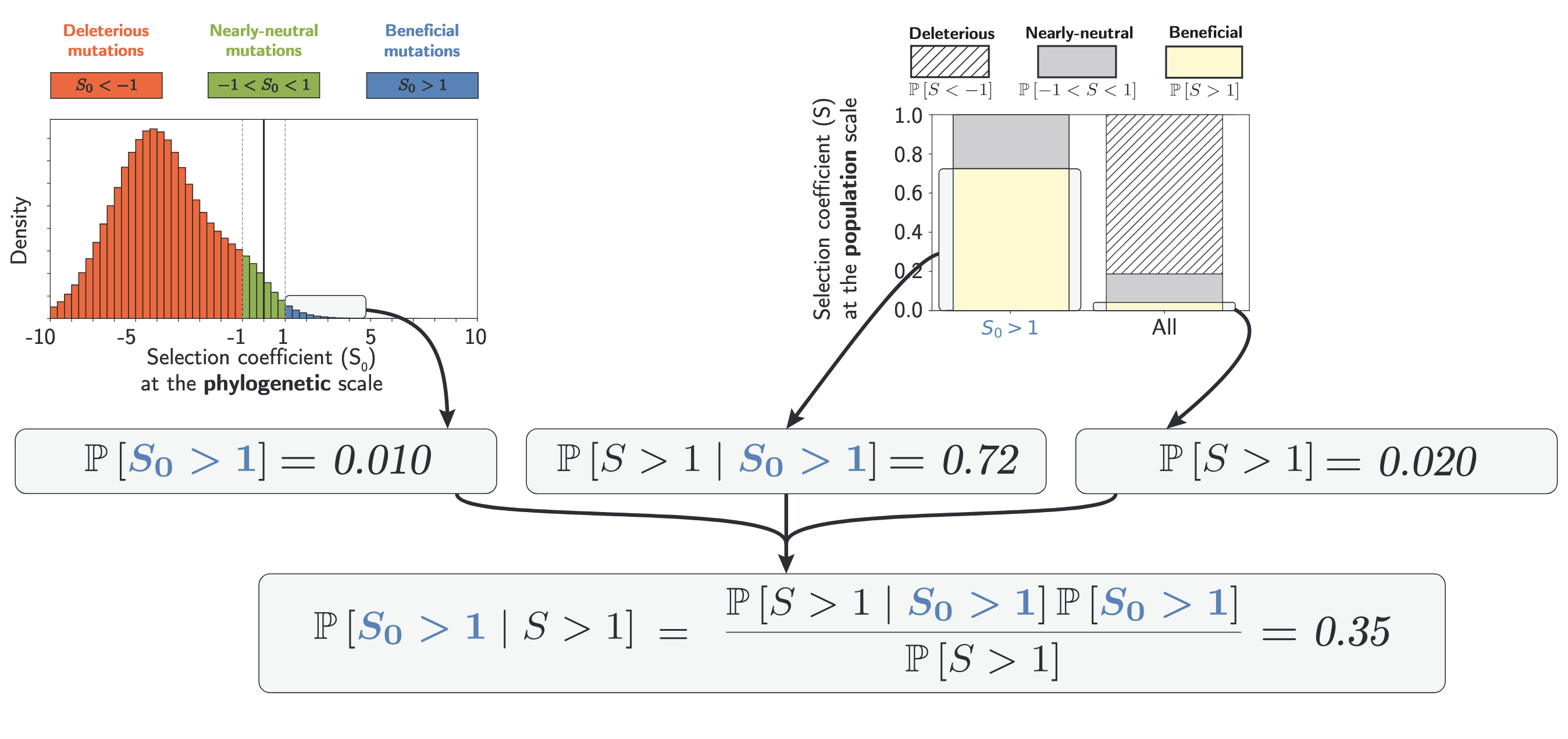
Using exomes of 96 mammals with polymorphism data for 28 populations from 6 genera, we show that back-mutations constitute an important fraction of beneficial mutations (P[B₀|B]).