Trait evolution across scales
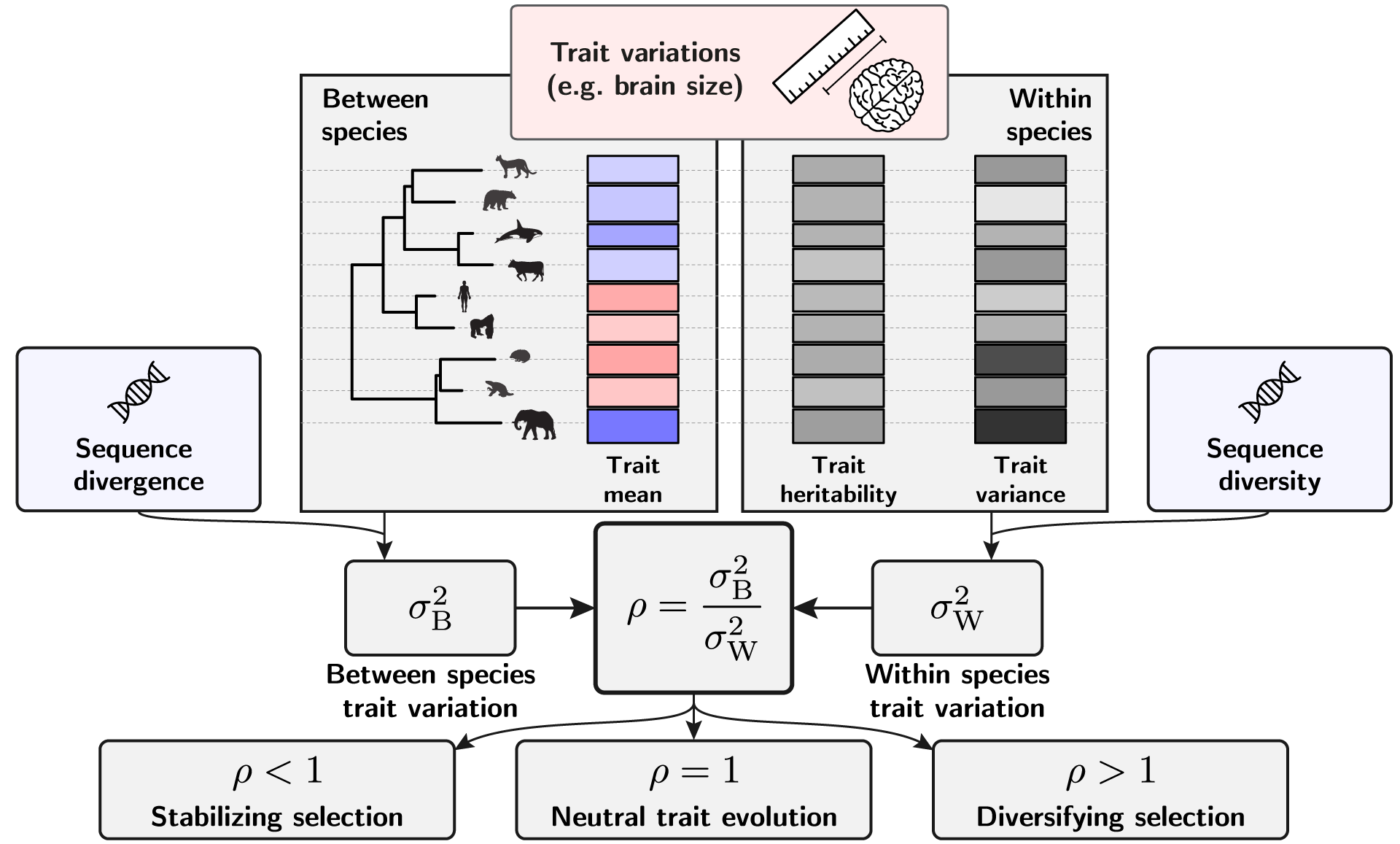
We proposed a neutrality index for a quantitative trait that can be used within a statistical framework to test for selection. Our neutrality index (ρ) is calculated as the ratio of the normalized between- to within-species variation. At the phylogenetic scale, trait variation between species was normalized by sequence divergence obtained from a neutral set of markers. Similarly, trait variation within species was normalized by sequence polymorphism obtained from a neutral set of markers.
We tested our statistical procedure against simulated data and showed that our test was able to correctly detect simulations under diversifying selection (ρ > 1) or under stabilizing selection (ρ < 1).
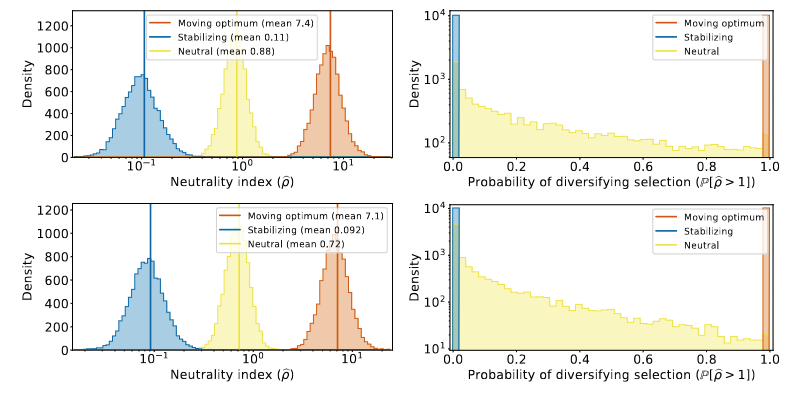
However, our test detected a spurious signal of stabilizing selection (ρ < 1) when we simulated the evolution of a neutral trait. Our test should be used to detect diversifying selection, but it had low accuracy to detect stabilizing selection due to false positives.
From an empirical point of view, our method required integrating genomic and trait variation, which could reduce the possible datasets to be used. However, such datasets will become more and more accessible and we showed the applicability of our method by applying it to the illustrative example of mammals brain and body mass.
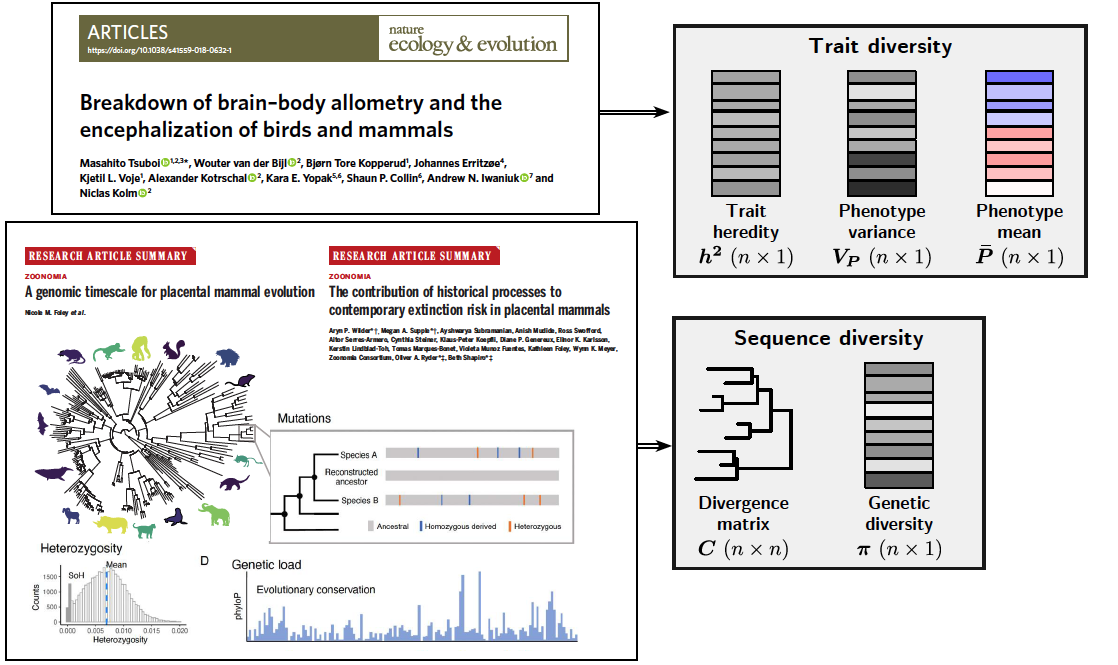
For geneticists used to detect selection for a protein-coding gene, our test can be seen as analogous to the $d_N / d_S$ ratio, but for a quantitative trait.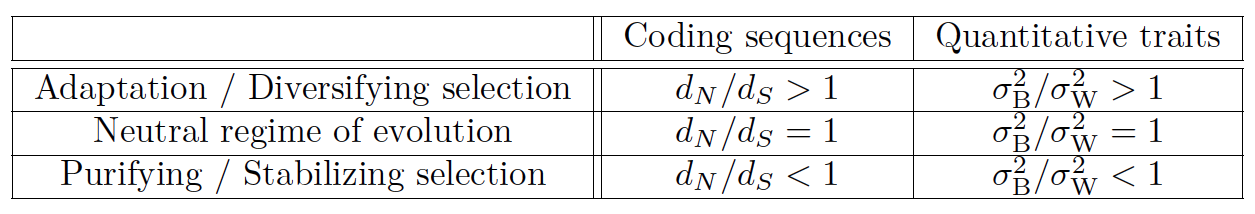
Our study bears similarities with others’ tests of selection. For example, our study can be seen as the macro-evolutionary analog of $Q_{ST}–F_{ST}$ methods to account for phylogenetic relationships between species.

From the field of phylogenetics, our study can be seen as an alternative to the EVE model (https://doi.org/10.1093/sysbio/syv042) for a single trait, where we set a threshold for neutral evolution by leveraging species nucleotide polymorphism and divergence.