Changing effects of mutations
Do the same mutations always have the same fitness effect?
Let’s study this question in the context of protein evolution, for which the protein folding is under selection, and the fitness effects of mutations are determined by their impact on folding stability. The question is whether the same mutation will have the same fitness effect regardless of the current stability of the protein. We can imagine that mutation that adds another destabilizing substitution to an already unstable protein will be far more costly than the same mutation on a highly stable protein. Let’s see how this intuition can be formalized and what the consequences are for the rate of protein evolution.
A natural model for the genotype-phenotype-fitness map of a protein is provided by folding thermodynamics. The protein-coding sequence determines its folding free energy $\Delta G = G_F - G_U$, where $G_F$ and $G_U$ are the free energies of the folded and unfolded states respectively. The fitness of the organism is assumed to proportional to the fraction of correctly folded proteins at any given moment, which increases steeply as $\Delta G$ becomes more negative (i.e. more stable) and saturates near one. An example can be due to the toxicity of misfolded proteins, which can aggregate and cause cellular damage.
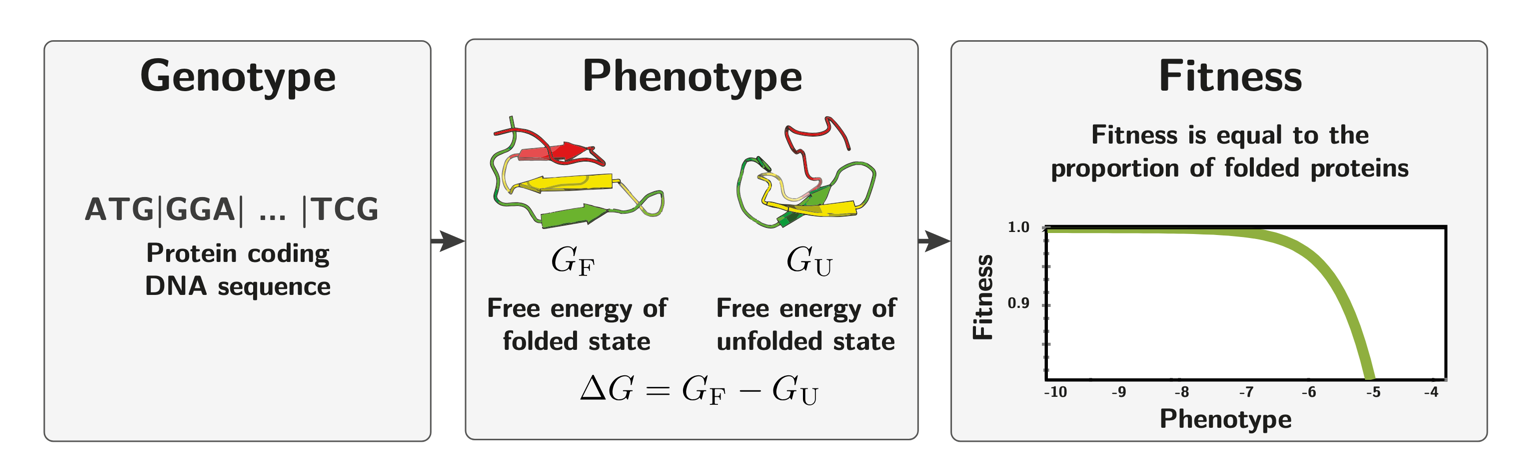
At mutation-selection equilibrium, the protein is marginally stable. This can be understood by a balance-of-forces argument on the fitness landscape. When the protein is highly stable (very negative $\Delta G$), most new mutations are destabilizing, but their selection coefficients are weak because the fitness function is nearly flat in the high-stability region. Consequently, destabilizing mutations drift toward fixation almost as readily as neutral ones. When the protein is unstable (less negative $\Delta G$), stabilizing mutations are strongly favoured by selection. The mutation-selection equilibrium therefore resides near the inflection point of the fitness curve (i.e. marginally stable) where the flux of destabilizing mutations arriving by mutation is exactly counterbalanced by the restoring force of selection against them.

The position of this equilibrium depends on the effective population size $N_e$. The reason is that $N_e$ determines the efficiency of natural selection, when $N_e$ increases, purifying selection against destabilizing mutations becomes more effective, and the equilibrium shifts toward a more stable protein (more negative $\Delta G$). This shift has a further consequence: because the log-fitness curve is concave, the local slope (which is the selection coefficient acting on any individual mutation at that protein site) changes as the equilibrium moves. An increase in $N_e$ thus not only stabilizes the protein but simultaneously decreases the magnitude of selection coefficients for any given mutation. The distribution of fitness effects at a site is therefore not a fixed property of the protein; it is coupled to $N_e$ through the equilibrium position.
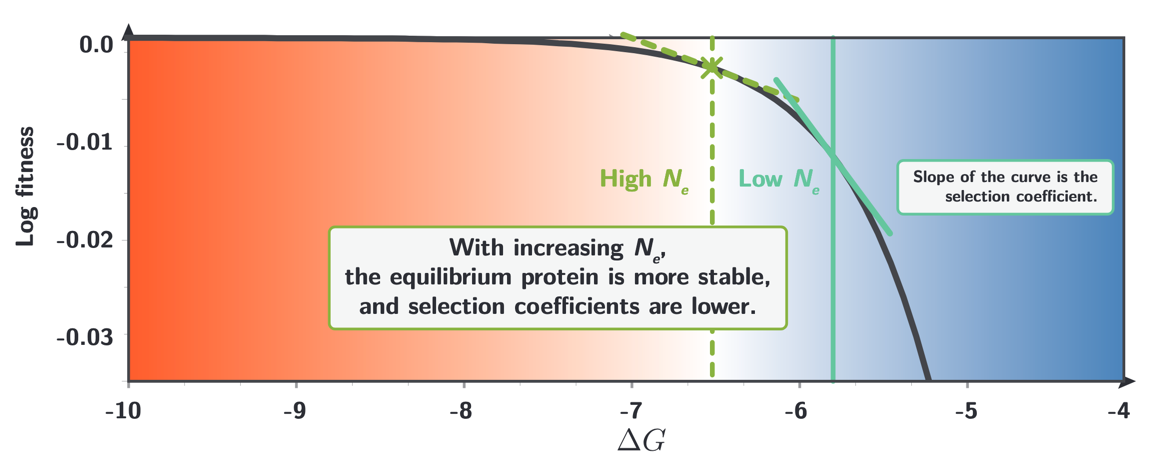
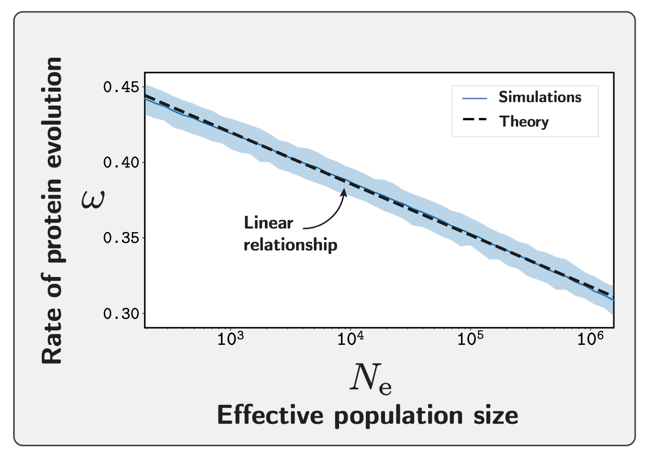
This coupling between $N_e$ and the rate of protein evolution $\omega$ can be made quantitative. Combining the statistical physics of the folding partition function with population genetics, the analytical approximation of the change in $\omega$ in response to a change in $N_e$ (in log scale) is linear and negative.
Tangentially, the same logic applies to other parameters that affect the position of the equilibrium. For example, protein expression level is known to correlate negatively with evolutionary rate $\omega$ (Drummond et al. 2005). This can be explained by the same model: if the protein is expressed at a higher level, the fitness cost of misfolded proteins increases, which steepens the fitness curve and shifts the equilibrium toward a more stable protein. Formally, the response of $\omega$ to a change in expression level is actually the same as the response to a change in $N_e$.

Individual-based simulations validate this approximation. Across several orders of magnitude in $N_e$, the rate of protein evolution $\omega$ decreases linearly with $\ln N_e$, with the predicted slope matching the simulations closely throughout.
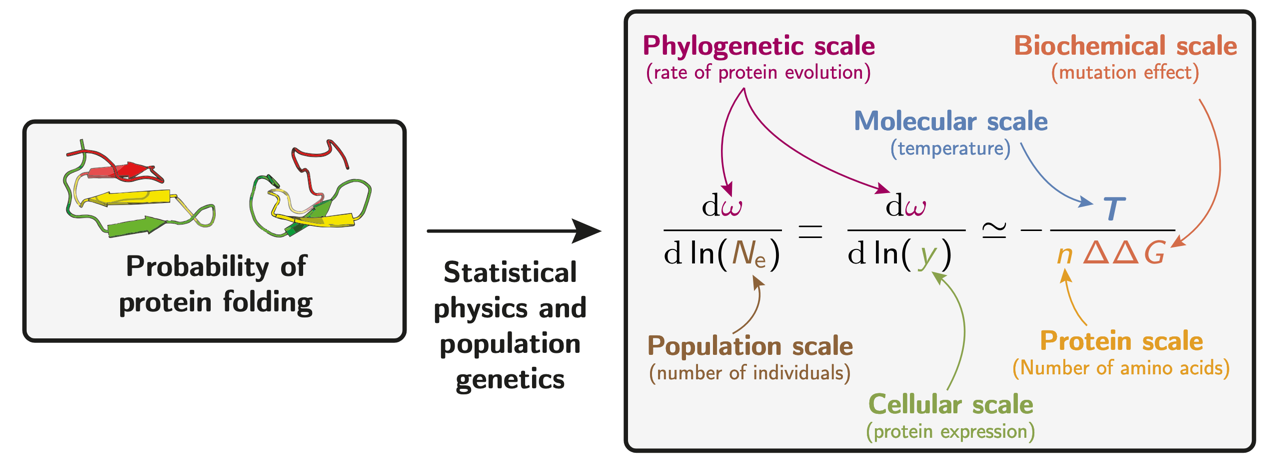
An unintended consequence of this model is the range of scales it connects. The probability of protein folding, combined with statistical physics and population genetics, links the phylogenetically observed rate $\omega$ to parameters at multiple biological levels: effective population size $N_e$ (population scale), temperature $T$ (molecular scale), protein expression level (cellular scale), the number of amino acids $n$ (protein scale), and the mean destabilizing effect $\Delta\Delta G$ (biochemical scale). A change in any one of these quantities shifts the mutation-selection equilibrium and the entire distribution of fitness effects at every protein site.
In summary, selection coefficients for protein-stability mutations are not fixed properties of the mutations themselves but emergent quantities that depend on the current position in the fitness landscape, which is itself set by $N_e$ and expression level. But here we assumed a stable fitness landscape, where the optimal folding stability does not change over time. In reality, the fitness landscape is likely to be dynamic, with the optimal stability shifting due to changes in the cellular environment, interactions with other proteins, or other evolutionary forces. This is discussed further in the context of phylogenetic inference in notes on mutation-selection models.